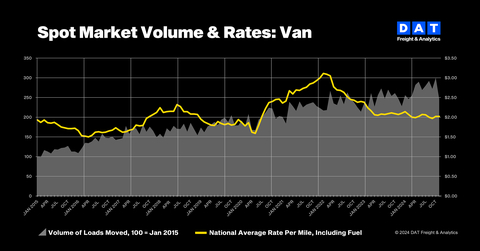
U.S. FDA Approves Pfizer’s BRAFTOVI® Combination Regimen as First-Line Treatment of BRAF V600E-Mutant Metastatic Colorectal Cancer
- BRAFTOVI in combination with cetuximab and mFOLFOX6 is the first and only combination regimen with targeted therapy approved for use as early as first-line for patients with metastatic colorectal cancer with a BRAFV600E mutation
- Accelerated approval is based on 61% overall response rate compared to 40% in control arm in the Phase 3 BREAKWATER trial
NEW YORK–(BUSINESS WIRE)–
Pfizer Inc. (NYSE: PFE) today announced that the U.S. Food and Drug Administration (FDA) has approved BRAFTOVI® (encorafenib) in combination with cetuximab (marketed as ERBITUX®) and mFOLFOX6 (fluorouracil, leucovorin, and oxaliplatin) for the treatment of patients with metastatic colorectal cancer (mCRC) with a BRAF V600E mutation, as detected by an FDA-approved test.i The indication was approved based on a statistically significant and clinically meaningful improvement in response rate and durability of response in treatment-naïve patients treated with BRAFTOVI in combination with cetuximab and mFOLFOX6 from the Phase 3 BREAKWATER trial. Continued approval for this indication is contingent upon verification of clinical benefit. This accelerated approval is among the first in the industry to be conducted under the FDA’s Project FrontRunner, which seeks to support the development and approval of new cancer drugs for advanced or metastatic disease.
“Historically, treatment options have been limited and outcomes poor for patients diagnosed with metastatic colorectal cancer with BRAF mutations,” said Scott Kopetz, M.D., Ph.D., FACP, Professor and Deputy Chair of Gastrointestinal Medical Oncology at The University of Texas MD Anderson Cancer Center and co-principal investigator of the BREAKWATER trial. “As the first and only combination regimen featuring a BRAF-targeted therapy for this patient population, usable even in first-line treatment, the encorafenib regimen has demonstrated high response rates that are rapid and durable. This represents an encouraging sign of continued disease control and a source of renewed hope for patients.”
The ongoing BREAKWATER trial is evaluating BRAFTOVI plus cetuximab with or without chemotherapy (mFOLFOX6) in previously untreated mCRC patients who harbor a BRAF V600E mutation. It is the only Phase 3 trial for a BRAF-targeted therapy regimen in first-line BRAF V600E-mutant mCRC. The trial met one of the dual primary endpoints of confirmed overall response rate (ORR), with a statistically significant improvement over standard of care: ORR 61% (95% confidence interval [CI]: 52, 70) for BRAFTOVI in combination with cetuximab and mFOLFOX6 compared with ORR 40% (95% CI: 31, 49) for chemotherapy, with or without bevacizumab (p=0.0008).1 The median duration of response (DoR) was 13.9 months (95% CI: 8.5, not estimable) for the BRAFTOVI combination regimen versus 11.1 months (95% CI: 6.7, 12.7) for chemotherapy, with or without bevacizumab.1 The Phase 3 BREAKWATER trial is ongoing, and results from the full dataset will be presented at upcoming medical meetings.
“For more than a decade, Pfizer has been a pioneer in delivering targeted therapies for molecular-driven cancers. With today’s accelerated approval of the BRAFTOVI regimen, patients with metastatic colorectal cancer with a BRAF V600E mutation now have a first-line treatment option, which contains a targeted therapy specifically for a mutation that is driving their cancer,” said Chris Boshoff, M.D., Ph.D., Chief Oncology Officer, Executive Vice President, Pfizer. “This milestone adds to our legacy of developing innovative medicines in BRAF tumors, some of the hardest-to-treat cancers. We look forward to continuing to expand our portfolio, including the exploration of a next-generation brain-penetrant BRAF inhibitor.”
The safety profile of BRAFTOVI in combination with cetuximab and mFOLFOX6 in the BREAKWATER trial was consistent with the known safety profile of each respective agent. No new safety signals were identified. The most common adverse reactions (≥25%) were peripheral neuropathy, nausea, fatigue, rash, diarrhea, decreased appetite, vomiting, hemorrhage, abdominal pain, and pyrexia.1 Among patients receiving BRAFTOVI in combination with cetuximab and mFOLFOX6, 12% experienced an adverse reaction that resulted in permanent discontinuation of BRAFTOVI; the most common adverse reactions (≥1%) included elevated lipase levels.
“Finding out that your cancer has spread can be a frightening time for those with colorectal cancer and their loved ones. The prognosis for those receiving a metastatic colorectal cancer diagnosis has improved slightly in recent years, but the same cannot be said for those with a BRAF mutation who unfortunately face an especially aggressive disease and worse outcomes,” said Michael Sapienza, Chief Executive Officer of the Colorectal Cancer Alliance. “Today’s approval of the first combination regimen including a BRAF-targeted therapy for BRAFV600E-mutant metastatic colorectal cancer, which can be used for previously untreated patients, offers new promise for the community and marks an important step forward in our collective mission to end this disease.”
This application was granted priority review, used the Real-Time Oncology Review (RTOR) pilot program, and was conducted under Project Orbis, with application reviews ongoing for Project Orbis partners, including Canada and Brazil. The BREAKWATER data are also being discussed with other regulatory authorities around the world to support potential future additional license applications for the BRAFTOVI combination regimen in this indication. This accelerated approval follows the previous FDA approval for BRAFTOVI in combination with cetuximab for adults with mCRC with a BRAF V600E mutation after prior therapy.
About Colorectal Cancer (CRC)
CRC is the third most common type of cancer in the world, with approximately 1.8 million new diagnoses in 2022.2 Overall, the lifetime risk of developing CRC is about 1 in 23 for men and 1 in 25 for women.3 In the U.S. alone, an estimated 152,810 people will be diagnosed with cancer of the colon or rectum in 2024, and approximately 53,000 are estimated to die from the disease each year.4 For 20% of those diagnosed with CRC, the disease has metastasized, or spread, making it harder to treat.5
BRAF mutations are estimated to occur in 8-10% of people with mCRC and represent a poor prognosis for these patients.6 The BRAF V600E mutation is the most common BRAF mutation and the risk of mortality in CRC patients with the BRAF V600E mutation is more than double that of patients with no known mutation present.6,7 Despite the high unmet need in BRAF V600E-mutant mCRC, prior to today there were no approved biomarker-driven therapies specifically indicated for people with previously untreated BRAF V600E-mutant mCRC.8,9
About BREAKWATER
BREAKWATER is a Phase 3, randomized, active-controlled, open-label, multicenter trial of BRAFTOVI with cetuximab, alone or in combination with chemotherapy, in participants with previously untreated BRAF V600E-mutant mCRC. In the Phase 3 portion of the study, patients were randomized to receive BRAFTOVI 300 mg orally once-daily in combination with cetuximab (discontinued after randomization of 158 patients), BRAFTOVI 300 mg orally once-daily in combination with cetuximab and mFOLFOX6 (n=236), or mFOLFOX6, FOLFOXIRI, or CAPOX each with or without bevacizumab (control arm) (n=243).
The dual primary endpoints are ORR and progression-free survival (PFS), as assessed by blinded independent central review (BICR). Key secondary endpoints include DoR as assessed by BICR, time to response by BICR, overall survival, and safety.
About BRAFTOVI® (encorafenib)
BRAFTOVI is an oral small molecule kinase inhibitor that targets BRAF V600E. Inappropriate activation of proteins in the MAPK signaling pathway (RAS-RAF-MEK-ERK) has been shown to occur in certain cancers, including CRC.
Pfizer has exclusive rights to BRAFTOVI in the U.S., Canada, Latin America, Middle East, and Africa. Ono Pharmaceutical Co., Ltd. has exclusive rights to commercialize the product in Japan and South Korea, Medison has exclusive rights to commercialize the product in Israel and Pierre Fabre has exclusive rights to commercialize the product in all other countries, including Europe and Asia (excluding Japan and South Korea).
U.S. INDICATION AND USAGE
BRAFTOVI® (encorafenib) is indicated, in combination with cetuximab and mFOLFOX6, for the treatment of patients with metastatic colorectal cancer (mCRC) with a BRAF V600E mutation, as detected by an FDA-approved test. This indication is approved under accelerated approval based on response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
BRAFTOVI is indicated, in combination with cetuximab, for the treatment of adult patients with mCRC with a BRAF V600E mutation, as detected by an FDA-approved test, after prior therapy.
Limitations of Use: BRAFTOVI is not indicated for treatment of patients with wild-type BRAF CRC.
IMPORTANT SAFETY INFORMATION
Refer to the prescribing information for cetuximab and individual product components of mFOLFOX6 for recommended dosing and additional safety information.
WARNINGS AND PRECAUTIONS
New Primary Malignancies: New primary malignancies, cutaneous and non-cutaneous, can occur. In BEACON CRC (previously treated BRAF V600E mutation-positive mCRC), cutaneous squamous cell carcinoma (cuSCC), including keratoacanthoma (KA), occurred in 1.4% of patients with CRC, and a new primary melanoma occurred in 1.4% of patients who received BRAFTOVI in combination with cetuximab. In BREAKWATER (previously untreated BRAF V600E mutation-positive mCRC) skin papilloma was reported in 2.6%, basal cell carcinoma in 1.3%, squamous cell carcinoma of skin in 0.9%, keratoacanthoma in 0.4% and malignant melanoma in situ in 0.4% of patients who received BRAFTOVI in combination with cetuximab and mFOLFOX6. Perform dermatologic evaluations prior to initiating treatment, every 2 months during treatment, and for up to 6 months following discontinuation of treatment. Manage suspicious skin lesions with excision and dermatopathologic evaluation. Dose modification is not recommended for new primary cutaneous malignancies. Based on its mechanism of action, BRAFTOVI may promote malignancies associated with activation of RAS through mutation or other mechanisms. Monitor patients receiving BRAFTOVI for signs and symptoms of non-cutaneous malignancies. Discontinue BRAFTOVI for RAS mutation-positive non-cutaneous malignancies. Monitor patients for new malignancies prior to initiation of treatment, while on treatment, and after discontinuation of treatment.
Tumor Promotion in BRAF Wild-Type Tumors: In vitro experiments have demonstrated paradoxical activation of MAP-kinase signaling and increased cell proliferation in BRAF wild-type cells exposed to BRAF inhibitors. Confirm evidence of BRAF V600E or V600K mutation using an FDA-approved test prior to initiating BRAFTOVI.
Cardiomyopathy: Cardiomyopathy manifesting as left ventricular dysfunction associated with symptomatic or asymptomatic decreases in ejection fraction, has been reported in patients. Assess left ventricular ejection fraction (LVEF) by echocardiogram or multi-gated acquisition (MUGA) scan prior to initiating treatment, 1 month after initiating treatment, and then every 2 to 3 months during treatment. The safety has not been established in patients with a baseline ejection fraction that is either below 50% or below the institutional lower limit of normal (LLN). Patients with cardiovascular risk factors should be monitored closely. Withhold, reduce dose, or permanently discontinue based on severity of adverse reaction.
Hepatotoxicity: Hepatotoxicity can occur. In BREAKWATER (previously untreated BRAF V600E mutation-positive mCRC), the incidence of Grade 3 or 4 increases in liver function laboratory tests in patients receiving BRAFTOVI in combination with cetuximab and mFOLFOX6 was 2.2% for alkaline phosphatase, 1.3% for ALT, and 0.9% for AST. Monitor liver laboratory tests before initiation of BRAFTOVI, monthly during treatment, and as clinically indicated. Withhold, reduce dose, or permanently discontinue based on severity of adverse reaction.
Hemorrhage: In BEACON CRC (previously treated BRAF V600E mutation-positive mCRC), hemorrhage occurred in 19% of patients receiving BRAFTOVI in combination with cetuximab; Grade 3 or higher hemorrhage occurred in 1.9% of patients, including fatal gastrointestinal hemorrhage in 0.5% of patients. The most frequent hemorrhagic events were epistaxis (6.9%), hematochezia (2.3%), and rectal hemorrhage (2.3%).In BREAKWATER (previously untreated BRAF V600E mutation-positive mCRC), hemorrhage occurred in 30% of patients receiving BRAFTOVI in combination with cetuximab and mFOLFOX6; Grade 3 or 4 hemorrhage occurred in 3% of patients. Withhold, reduce dose, or permanently discontinue based on severity of adverse reaction.
Uveitis: Uveitis, including iritis and iridocyclitis, has been reported in patients treated with BRAFTOVI. Assess for visual symptoms at each visit. Perform an ophthalmological evaluation at regular intervals and for new or worsening visual disturbances, and to follow new or persistent ophthalmologic findings. Withhold, reduce dose, or permanently discontinue based on severity of adverse reaction.
QT Prolongation: BRAFTOVI is associated with dose-dependent QTc interval prolongation in some patients. In BREAKWATER (previously untreated BRAF V600E mutation-positive mCRC), an increase of QTcF >500 ms was measured in 3.6% (8/222) of patients receiving BRAFTOVI in combination with cetuximab and mFOLFOX6. Monitor patients who already have or who are at significant risk of developing QTc prolongation, including patients with known long QT syndromes, clinically significant bradyarrhythmias, severe or uncontrolled heart failure and those taking other medicinal products associated with QT prolongation. Correct hypokalemia and hypomagnesemia prior to and during BRAFTOVI administration. Withhold, reduce dose, or permanently discontinue for QTc >500 ms.
Embryo-Fetal Toxicity: BRAFTOVI can cause fetal harm when administered to pregnant women. BRAFTOVI can render hormonal contraceptives ineffective. Advise females of reproductive potential to use effective nonhormonal contraception during treatment with BRAFTOVI and for 2 weeks after the final dose.
Risks Associated with Combination Treatment: BRAFTOVI is indicated for use as part of a regimen in combination with cetuximab, or in combination with cetuximab and mFOLFOX6. Refer to the prescribing information for cetuximab and individual product components of mFOLFOX6 for additional risk information.
Lactation: Advise women not to breastfeed during treatment with BRAFTOVI and for 2 weeks after the final dose.
Infertility: Advise males of reproductive potential that BRAFTOVI may impair fertility.
ADVERSE REACTIONS
BREAKWATER Trial (previously untreated BRAF V600E mutation-positive mCRC)
- Serious adverse reactions occurred in 38% of patients who received BRAFTOVI in combination with cetuximab and mFOLFOX6. Serious adverse reactions in >3% of patients included intestinal obstruction (3.5%) and pyrexia (3.5%).
- Fatal gastrointestinal perforation occurred in 0.9% of patients who received BRAFTOVI in combination with cetuximab and mFOLFOX6.
- Most common adverse reactions (≥25%, all grades) in the BRAFTOVI with cetuximab and mFOLFOX6 arm compared to the control arm (mFOLFOX6 ± bevacizumab or FOLFOXIRI ± bevacizumab or CAPOX ± bevacizumab) were peripheral neuropathy (62% vs 53%), nausea (51% vs 48%), fatigue (49% vs 38%), rash (31% vs 4%), diarrhea (34% vs 47%), decreased appetite (33% vs 25%), vomiting (33% vs 21%), hemorrhage (30% vs 18%), abdominal pain (26% vs 27%), and pyrexia (26% vs 14%).
- Most common laboratory abnormalities (≥10%, grade 3 or 4) in the BRAFTOVI with cetuximab and mFOLFOX6 arm compared to the control arm (mFOLFOX6 ± bevacizumab or FOLFOXIRI ± bevacizumab or CAPOX ± bevacizumab) were: increased lipase (51% vs 25%), decreased neutrophil count (36% vs 34%), decreased hemoglobin (13% vs 5%), decreased white blood cell count (12% vs 7%), and increased glucose (11% vs 2%).
BEACON CRC Trial (previously treated BRAF V600E mutation-positive mCRC)
- Most common adverse reactions (≥25%, all grades) in the BRAFTOVI with cetuximab arm compared to irinotecan with cetuximab or FOLFIRI with cetuximab (control) were: fatigue (51% vs 50%), nausea (34% vs 41%), diarrhea (33% vs 48%), dermatitis acneiform (32% vs 43%), abdominal pain (30% vs 32%), decreased appetite (27% vs 27%), arthralgia (27% vs 3%), and rash (26% vs 26%).
- Other clinically important adverse reactions occurring in <10% of patients who received BRAFTOVI in combination with cetuximab was pancreatitis.
- Most common laboratory abnormalities (all grades) (≥20%) in the BRAFTOVI with cetuximab arm compared to irinotecan with cetuximab or FOLFIRI with cetuximab (control) were: anemia (34% vs 48%) and lymphopenia (24% vs 35%).
DRUG INTERACTIONS
Strong or moderate CYP3A4 inhibitors: Avoid coadministration of BRAFTOVI with strong or moderate CYP3A4 inhibitors, including grapefruit juice. If coadministration is unavoidable, reduce the BRAFTOVI dose.
Strong CYP3A4 inducers: Avoid coadministration of BRAFTOVI with strong CYP3A4 inducers.
Sensitive CYP3A4 substrates: Avoid the coadministration of BRAFTOVI with CYP3A4 substrates (including hormonal contraceptives) for which a decrease in plasma concentration may lead to reduced efficacy of the substrate. If the coadministration cannot be avoided, see the CYP3A4 substrate product labeling for recommendations.
Dose reductions of drugs that are substrates of OATP1B1, OATP1B3, or BCRP may be required when used concomitantly with BRAFTOVI.
Avoid coadministration of BRAFTOVI with drugs known to prolong QT/QTc interval.
View the full Prescribing Information. There may be a delay as the document is updated with the latest information. It will be available as soon as possible. Please check back for the updated full information shortly.
About Pfizer Oncology
At Pfizer Oncology, we are at the forefront of a new era in cancer care. Our industry-leading portfolio and extensive pipeline includes three core mechanisms of action to attack cancer from multiple angles, including small molecules, antibody-drug conjugates (ADCs), and bispecific antibodies, including other immune-oncology biologics. We are focused on delivering transformative therapies in some of the world’s most common cancers, including breast cancer, genitourinary cancer, hematology-oncology, and thoracic cancers, which includes lung cancer. Driven by science, we are committed to accelerating breakthroughs to help people with cancer live better and longer lives.
About Pfizer: Breakthroughs That Change Patients’ Lives
At Pfizer, we apply science and our global resources to bring therapies to people that extend and significantly improve their lives. We strive to set the standard for quality, safety and value in the discovery, development and manufacture of health care products, including innovative medicines and vaccines. Every day, Pfizer colleagues work across developed and emerging markets to advance wellness, prevention, treatments and cures that challenge the most feared diseases of our time. Consistent with our responsibility as one of the world’s premier innovative biopharmaceutical companies, we collaborate with health care providers, governments and local communities to support and expand access to reliable, affordable health care around the world. For 175 years, we have worked to make a difference for all who rely on us. We routinely post information that may be important to investors on our website at www.Pfizer.com. In addition, to learn more, please visit us on www.Pfizer.com and follow us on X at @Pfizer and @Pfizer_News, LinkedIn, YouTube and like us on Facebook at Facebook.com/Pfizer.
Disclosure Notice
The information contained in this release is as of December 20, 2024. Pfizer assumes no obligation to update forward-looking statements contained in this release as the result of new information or future events or developments.
This release contains forward-looking information about the BRAFTOVI® (encorafenib) plus cetuximab and mFOLFOX6 combination and a new indication in the U.S. for thetreatment of patients with metastatic colorectal cancer (CRC) with a BRAF V600E mutation, as detected by an FDA-approved test, including their potential benefits and discussions with other regulatory authorities to support potential future additional license applications for the BRAFTOVI combination regimen in this indication, that involves substantial risks and uncertainties that could cause actual results to differ materially from those expressed or implied by such statements. Risks and uncertainties include, among other things, uncertainties regarding the commercial success of BRAFTOVI plus cetuximab and mFOLFOX6; the uncertainties inherent in research and development, including the ability to meet anticipated clinical endpoints, commencement and/or completion dates for our clinical trials, regulatory submission dates, regulatory approval dates and/or launch dates, as well as the possibility of unfavorable new clinical data and further analyses of existing clinical data; the risk that clinical trial data are subject to differing interpretations and assessments by regulatory authorities; whether regulatory authorities will be satisfied with the design of and results from our clinical studies; whether and when any drug applications may be filed in any additional jurisdictions for BRAFTOVI plus cetuximab and mFOLFOX6 for the treatment of patients with metastatic CRC with a BRAF V600E mutation or in any jurisdictions for any other potential indications for BRAFTOVI; whether and when any such other applications may be approved by regulatory authorities, which will depend on a myriad factors, including making a determination as to whether the product’s benefits outweigh its known risks and determination of the product’s efficacy and, if approved, whether BRAFTOVI plus cetuximab and mFOLFOX6 will be commercially successful; decisions by regulatory authorities impacting labeling, manufacturing processes, safety and/or other matters that could affect the availability or commercial potential of BRAFTOVI or BRAFTOVIplus cetuximab and mFOLFOX6; uncertainties regarding the impact of COVID-19 on Pfizer’s business, operations and financial results; and competitive developments.
A further description of risks and uncertainties can be found in Pfizer’s Annual Report on Form 10-K for the fiscal year ended December 31, 2023, and in its subsequent reports on Form 10-Q, including in the sections thereof captioned “Risk Factors” and “Forward-Looking Information and Factors That May Affect Future Results”, as well as in its subsequent reports on Form 8-K, all of which are filed with the U.S. Securities and Exchange Commission and available at www.sec.gov and www.pfizer.com.
Erbitux® is a registered trademark of Eli Lilly and Company and Merck KGaA, Darmstadt, Germany.
References
______________________
1 BRAFTOVI (encorafenib) Prescribing Information. Array BioPharma, Inc.; December 2024
2 American Cancer Society. Global Cancer Facts & Figures 5th Edition. Available at: https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/global-cancer-facts-and-figures/global-cancer-facts-and-figures-2024.pdf. Last accessed: December 2024.
3 American Cancer Society. Key Statistics for Colorectal Cancer. Available at: https://www.cancer.org/cancer/types/colon-rectal-cancer/about/key-statistics.html. Last accessed: December 2024.
4 American Cancer Society. Cancer Facts & Figures 2024. Available at: https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2024/2024-cancer-facts-and-figures-acs.pdf. Last accessed: December 2024.
5 Ciardiello F, Ciardiello D, Martini G, et al. Clinical management of metastatic colorectal cancer in the era of precision medicine. CA Cancer J Clin. 2022;72:372–40.
6 Djanani A, Eller S, Öfner D, et al. The role of BRAF in metastatic colorectal carcinoma-past, present, and future. Int J Mol Sci. 2020;21(23):9001.
7 Safaee Ardekani G, Jafarnejad SM, Tan L, et al. The prognostic value of BRAF mutation in colorectal cancer and melanoma: a systematic review and meta-analysis. PloS ONE. 2012;7(10):e47054.
8 NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines) for Colon Cancer. V.5.2024 © National Comprehensive Cancer Network, Inc. 2024. All rights reserved. Accessed December 2024. To view the most recent and complete version of the guideline, go online to NCCN.org.
9 Cervantes A, Adam R, Roselló S, et al. Metastatic colorectal cancer: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann Oncol. 2023;34(1):10–32.
Category: Prescription Medicines

View source version on businesswire.com: https://www.businesswire.com/news/home/20241210321033/en/
Media Contact:
+1 (212) 733-1226
[email protected]
Investor Contact:
+1 (212) 733-4848
[email protected]
KEYWORDS: New York United States North America
INDUSTRY KEYWORDS: Research FDA Clinical Trials Biotechnology Health Pharmaceutical General Health Science Oncology
MEDIA:
| Logo |
 |
|